Snakemake workflow catalog
News
2026-04-13
We are excited to launch two new features in the Snakemake Workflow Catalog that make automatic display of workflow pages both easier and more visually appealing:
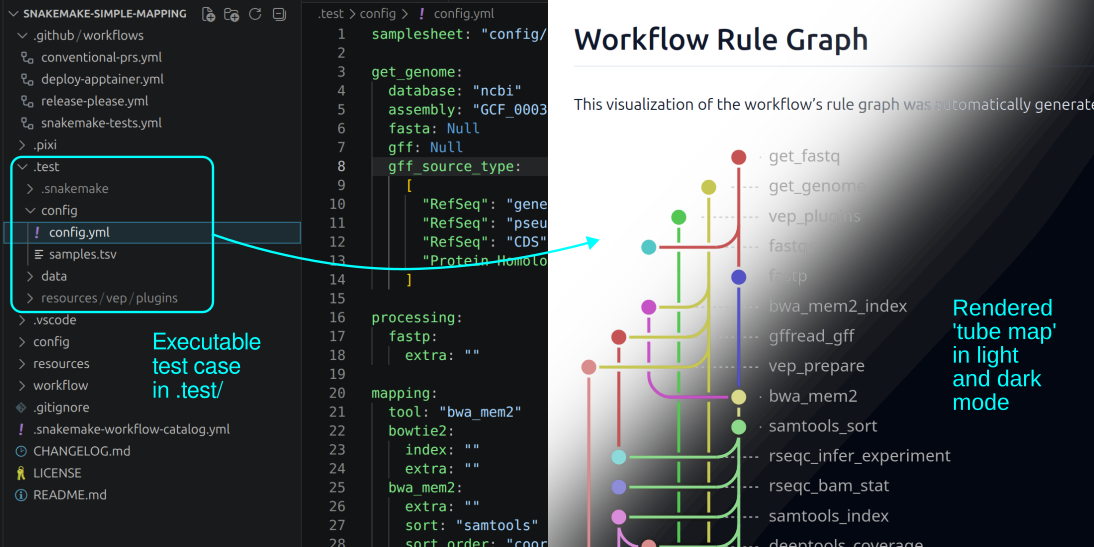
The Catalog now supports automatic rendering of Tube Maps using snakevision. Tube maps will automatically show up on your workflow page if the workflow has an executable test case in
.test(details).
The Catalog will automatically render a Workflow Parameter Table if your workflow repo contains a
workflow/schemas/config.schema.y(a)mlorconfig/schemas/config.schema.y(a)ml(details). No need to manually maintain a table of parameters on your workflow page anymore.

About
The Snakemake Workflow Catalog aims to provide a regularly updated list of high-quality workflows that can be easily reused and adapted for various data analysis tasks. By leveraging the power of Snakemake, these workflows promote:
Snakemake workflows produce consistent results, making it easier to share and validate scientific findings.
![]()
Snakemake workflows can be executed on various environments, from local machines to clusters and clouds.
![]()
Snakemake workflows allow easy customization and extension, enabling users to adapt them to their needs.
![]()
Short cuts
Workflows in numbers
Contributing
Improving PRs or issues with the workflow catalog (only the catalog, not the workflows themselves) can be made here
Improving PRs or issues with the listed workflows can be made at the respective workflow repository (see individual workflow pages).
Resources for creating new workflows can be found here or in more detail on the Snakemake documentation
New workflows will be automatically added to the workflow catalog if they are contained in eligible Github repositories